Sleep is the most underrated metabolic lever. A study from the University of Chicago found that when dieters were sleep-deprived (5.5 hours), they lost 55% less fat and felt significantly hungrier compared to when they slept 8.5 hours—even though their calorie intake was identical.
The Glymphatic System
During deep sleep, your brain’s glymphatic system opens up, allowing cerebrospinal fluid to wash away metabolic waste (including beta-amyloid). Depriving yourself of this “wash” leads to cognitive decline and increased cortisol levels.
The Sleep Optimization Protocol
1. The 10-3-2-1-0 Rule: 10 hours before bed: No caffeine. 3 hours: No food. 2 hours: No work. 1 hour: No screens. 0: Times you hit snooze.
2. Magnesium & Temperature: Keep your room at 18°C. Consider magnesium glycinate to support the nervous system.
3. Track Recovery: Use the [HRV Calculator](/hrv-calculator/) to see how your sleep quality affects your heart rate variability—the gold standard for recovery data.
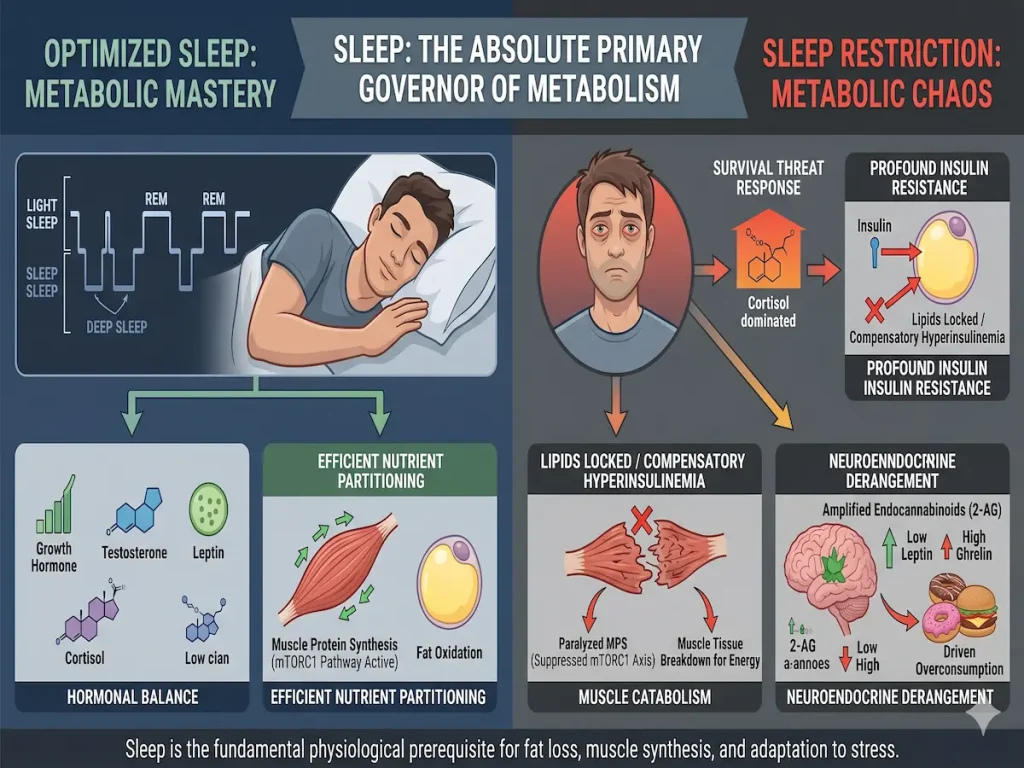
Sleep as a Systemic Performance Metric: The Neuroendocrine and Metabolic Consequences of Sleep Restriction
Introduction to Sleep Architecture and Metabolic Physiology
Historically conceptualized within the medical and athletic communities as a passive state of neurological rest, human sleep is increasingly understood by modern endocrinologists and metabolic researchers as an active, highly regulated, and immensely powerful metabolic lever. For decades, the prevailing dogma of body composition manipulation and human performance optimization rested almost exclusively on the thermodynamic principles of energy balance and mechanical training load. Under this reductionist framework, caloric deficits were universally assumed to drive lipolysis and weight loss, while caloric surpluses coupled with progressive resistance training were assumed to drive muscular hypertrophy. However, an exhaustive analysis of contemporary clinical literature demonstrates that the absolute quantity of ingested energy represents only a fraction of the physiological equation. The composition of the human body—specifically the critical ratio of metabolically active fat-free mass to stored adipose tissue—is heavily, and sometimes entirely, dictated by the neuroendocrine milieu present during the intervention.
Sleep architecture, which encompasses the precise cycling of slow-wave deep sleep, rapid eye movement (REM) phases, and total nocturnal sleep duration, exerts profound and instantaneous control over the physiological pathways responsible for systemic substrate utilization. When sleep is curtailed, whether through clinical intervention, social jet lag, occupational demands, or chronic psychological stress, the neuroendocrine system initiates an aggressive cascade of compensatory adaptations. These adaptations fundamentally alter how the human body metabolizes exogenous carbohydrates, synthesizes endogenous structural proteins, regulates adipose tissue stores, and perceives the reward value of hyperpalatable foods.
The characterization of sleep deprivation as merely a behavioral inconvenience that results in cognitive fatigue drastically underestimates its systemic pathology. At a molecular level, restricted sleep acts as a potent systemic stressor that disrupts endocrine homeostasis, promoting a distinctly catabolic environment within skeletal muscle while simultaneously inducing severe anabolic resistance and hyperinsulinemia in peripheral tissues. Consequently, sleep deprivation emerges as a primary pathogenic driver of metabolic syndrome, profound insulin resistance, and suboptimal athletic performance. This document provides a comprehensive, exhaustive analysis of the second- and third-order metabolic consequences of sleep restriction, detailing its impact on macronutrient partitioning, cellular insulin signaling, myofibrillar protein synthesis, the neurological pathways governing human appetite, and the integration of objective biometric quantification in modern sports science.
The University of Chicago Adiposity Trial: A Paradigm Shift in Nutrient Partitioning
The most compelling and paradigm-altering evidence demonstrating sleep’s role as a primary governor of body composition emerges from rigorous, tightly controlled clinical trials examining macronutrient partitioning under identical, strictly monitored caloric deficits. A landmark randomized, two-period, two-condition crossover study conducted at the University of Chicago’s General Clinical Resource Center systematically dismantled the long-standing assumption that caloric deficits yield uniform physiological outcomes independent of sleep duration.
The research protocol was meticulously designed to isolate sleep duration as the sole independent metabolic variable. The trial followed a cohort of ten overweight but otherwise healthy, nonsmoking adult volunteers. The participant demographics included three women and seven men with a mean age of 41 years (standard deviation of 5 years) and a mean body mass index (BMI) of $27.4 \, kg/m^2$ (standard deviation of 2.0), ranging from 25 (categorized as overweight) to 32 (categorized as obese). The participants were placed on a highly individualized, balanced macronutrient diet. Crucially, caloric intake was strictly restricted to exactly 90 percent of what each individual person required to maintain his or her baseline body weight without factoring in additional exercise. This equated to an average daily intake of approximately 1,450 kilocalories per participant.
Each participant was continuously monitored in a closed clinical laboratory environment for two distinct 14-day periods. During one phase, the participants were provided with an 8.5-hour period set aside specifically for sleep opportunity. During the cross-over phase, the sleep opportunity was strictly curtailed to only 5.5 hours per night. During their waking hours, the participants engaged in identical home- or office-like work and sedentary leisure activities to ensure energy expenditure remained stable across both testing periods.
The primary measure of the study was the assessment of compartmentalized weight loss—specifically distinguishing between the loss of fat mass and the loss of fat-free body mass (predominantly skeletal muscle and water)—assessed while absolute caloric intake remained perfectly static. During the 8.5-hour time-in-bed phase, volunteers objectively slept an average of 7 hours and 25 minutes each night. Conversely, during the 5.5-hour phase, they slept an average of 5 hours and 14 minutes, representing a sustained restriction of more than two hours of actual sleep per night.
The macroscopic weight loss observed during both 14-day hypocaloric sessions was nearly identical, with participants losing an average of 3.0 kilograms (approximately 6.6 pounds) during each phase. If one were to analyze the results purely from the perspective of the scale, the interventions would appear equally successful. However, dual-energy x-ray absorptiometry and metabolic analyses revealed that the compartmental source of that weight loss diverged dramatically, dictated entirely by the sleep variable.
| Metabolic Outcome Metric | 8.5-Hour Sleep Opportunity | 5.5-Hour Sleep Opportunity | Statistical Significance |
| Actual Sleep Achieved | 7 hours, 25 minutes | 5 hours, 14 minutes | N/A |
| Absolute Fat Loss | 1.4 kg ($\pm$ 0.9) | 0.6 kg ($\pm$ 0.6) | $P = 0.043$ |
| Proportion of Weight Lost as Fat | 56% ($\pm$ 35%) | 25% ($\pm$ 24%) | $P = 0.043$ |
| Absolute Fat-Free Mass Loss | 1.5 kg ($\pm$ 1.3) | 2.4 kg ($\pm$ 1.4) | $P = 0.002$ |
| Proportion of Weight Lost as Fat-Free Mass | 44% | 75% | $P = 0.002$ |
| Fasting Respiratory Quotient (RQ) | 0.80 ($\pm$ 0.04) | 0.83 ($\pm$ 0.04) | N/A |
| Postprandial RQ (4-hour mean) | 0.80 ($\pm$ 0.04) | 0.83 ($\pm$ 0.05) | N/A |
Analysis of macronutrient partitioning and body composition shifts under varying sleep durations with identical hypocaloric diets.
The results of this study fundamentally isolate sleep as an independent, highly potent metabolic lever. Curtailing sleep by roughly two hours per night decreased the proportion of weight lost as fat by an astonishing 55 percent, reducing the absolute fat loss from 1.4 kilograms down to a mere 0.6 kilograms. Under adequate sleep conditions, more than half of the weight lost (56 percent) was sourced from adipose tissue. Under restricted sleep, only one-fourth (25 percent) of the weight loss came from fat. Conversely, the loss of metabolically active fat-free body mass increased by 60 percent, jumping from 1.5 kilograms to 2.4 kilograms. Lead researchers analogized this physiological self-sabotage to introducing mechanical friction into a calibrated system, noting that if the goal of a dietary intervention is to reduce adiposity, skipping sleep is effectively like poking sticks into the spokes of a bicycle wheel.
The underlying physiological mechanisms driving this aggressive shift in nutrient partitioning are revealed by the participants’ Respiratory Quotient (RQ). The RQ is a dimensionless metabolic ratio calculated by comparing the volume of carbon dioxide produced to the volume of oxygen consumed. It is utilized to estimate the specific macronutrient mixture being oxidized for cellular energy. An RQ approaching 0.70 indicates near-pure lipid oxidation (fat burning), whereas an RQ approaching 1.0 indicates near-pure carbohydrate oxidation. Under the 5.5-hour sleep condition, the fasting RQ shifted measurably upward from 0.80 to 0.83, indicating a systemic, biologically mediated downregulation of beta-oxidation.
This upward shift in RQ implies that sleep restriction triggers a profound “starvation illusion” within the central nervous system. Despite identical exogenous caloric intake, the extended wakefulness, coupled with the neuroendocrine stress of sleep deprivation, signals an existential energetic crisis to the brain. The human body responds to this perceived crisis through highly conserved evolutionary mechanisms: it fiercely defends its calorically dense lipid stores to ensure long-term survival, while simultaneously upregulating the oxidation of circulating glucose and endogenous amino acids. Consequently, clinical efforts to lose adiposity through standard dietary interventions are profoundly undermined. The dieter inadvertently cannibalizes their own skeletal muscle architecture while simultaneously preserving the very visceral and subcutaneous adipose tissue they are actively attempting to shed. Sleep, therefore, is not merely an auxiliary component of a weight management program; it is the fundamental physiological prerequisite that permits the lipolytic mobilization of triglycerides in a hypocaloric state.
Peripheral Insulin Resistance and the Pathophysiology of Adipocyte Dysfunction
To fully grasp why systemic nutrient partitioning shifts so drastically away from lipid oxidation and toward structural tissue catabolism under sleep curtailment, a granular examination of cellular metabolic pathways is required. Adipose tissue is not a biologically inert storage depot, as it was historically conceptualized. Rather, it is a highly active, heavily vascularized, and acutely insulin-sensitive endocrine organ that dynamically secretes a host of cytokines, inflammatory markers, and adipokines (such as leptin and adiponectin) to regulate systemic energy balance and peripheral glucose uptake.
Short-term sleep deprivation rapidly induces a state of profound insulin resistance in peripheral tissues, specifically targeting the functional capacity and metabolic flexibility of subcutaneous adipocytes. A definitive, first-of-its-kind study isolating this phenomenon investigated a cohort of seven healthy, lean men and women (aged 18 to 30) who were evaluated in a sleep laboratory. In a randomized crossover design, participants underwent four nights of restricted sleep (4.5 hours in bed) compared to four nights of adequate sleep (8.5 hours in bed), with a washout period of at least four weeks between conditions. Physical activity and caloric intake were strictly controlled to eliminate confounding variables regardless of sleep duration.
At the conclusion of each study period, researchers administered an intravenous glucose-tolerance test to measure total-body insulin sensitivity, followed immediately by a biopsy of abdominal subcutaneous fat cells to measure how the individual adipocytes responded to insulin exposure at the molecular level. The biopsies specifically measured the phosphorylation of Akt (protein kinase B), which is an essential, early, and rate-limiting chemical step in the cellular insulin-signaling cascade. Under normal physiological conditions, when insulin binds to its extracellular receptor on the adipocyte membrane, it triggers a cascade involving insulin receptor substrate 1 (IRS-1) and phosphoinositide 3-kinase (PI3K), which ultimately results in the phosphorylation of Akt (pAkt). Phosphorylated Akt then facilitates the translocation of GLUT4 transport proteins from intracellular vesicles to the cell membrane, allowing the cell to rapidly ingest circulating plasma glucose. The ratio of phosphorylated Akt to total Akt (pAkt/tAkt) serves as a direct, highly sensitive, and objective proxy for cellular insulin sensitivity.
Following just four nights of sleep restriction, the total-body insulin response decreased by an average of 16 percent. However, the localized, tissue-specific insulin sensitivity of the biopsied adipocytes plummeted by an astonishing 30 percent. To provoke half of the maximum Akt phosphorylation response, the sleep-deprived adipocytes required nearly three times as much exogenous insulin. The magnitude of this 30 percent reduction in cellular insulin signaling is clinically severe; researchers explicitly noted that this level of impairment effectively degrades the metabolic functioning of a healthy, young, lean individual’s fat cells to a state that is statistically indistinguishable from the adipocytes of an obese patient, or a patient suffering from established, chronic type 2 diabetes. Strikingly, every single subject in the trial (seven out of seven) demonstrated this significant molecular derangement, indicating that metabolic dysfunction is a universal physiological response to sleep deprivation, rather than an individualized vulnerability.
The impairment of adipocyte insulin signaling provides a direct, causal, and elegant molecular mechanism for the skewed nutrient partitioning observed during the University of Chicago adiposity trial. When adipocytes become profoundly insulin resistant, they lose their capacity to efficiently clear circulating postprandial glucose. This peripheral resistance initiates a dangerous systemic feedback loop: the pancreas is forced to secrete supraphysiological volumes of insulin to maintain euglycemia, resulting in compensatory hyperinsulinemia.
Chronically elevated circulating insulin levels fundamentally alter the cellular environment. Crucially, insulin is the most potent anti-lipolytic hormone in the human body. It actively suppresses and inhibits the enzyme hormone-sensitive lipase (HSL), which is responsible for cleaving stored intracellular triglycerides into free fatty acids and glycerol so they can be released into the bloodstream for beta-oxidation by peripheral tissues. Therefore, the hyperinsulinemia driven by sleep-deprivation-induced adipocyte resistance biochemically locks adipose stores, trapping lipids within the cell and halting fat loss at the source. This mechanism explains precisely why the sleep-restricted participants could not lose body fat despite being in a mathematically validated caloric deficit. Because the body cannot access its fat stores due to high circulating insulin, but still requires energy to meet the metabolic demands of an artificially extended waking period, it is forced to source energy elsewhere. This energetic triage leads directly to the accelerated catabolism of skeletal muscle tissue, breaking down functional proteins into their constituent amino acids to fuel hepatic gluconeogenesis.
Skeletal Muscle Integrity: Anabolic Resistance and Myofibrillar Protein Synthesis
Skeletal muscle mass is not a static tissue; it is highly plastic and regulated by the continuous, dynamic interplay between muscle protein synthesis (MPS) and muscle protein breakdown (MPB). A net positive balance between these two processes yields muscular hypertrophy, while a net negative balance causes muscular atrophy. The aggressive loss of fat-free mass observed under sleep restriction is primarily driven not by an immediate, massive surge in proteolytic tissue destruction, but rather by an acute, catastrophic failure in the synthetic pathways, generating a state of severe anabolic resistance.
The direct relationship between sleep loss and skeletal muscle metabolism was rigorously quantified using advanced stable isotopic tracer methodologies in a randomized crossover trial. Healthy young adults (N=13; comprising seven males and six females) were subjected to a single night of total sleep deprivation compared to a normal night of control sleep. Deuterium oxide, alongside an infusion of the tracer [ring-13C6]-L-phenylalanine, was administered to precisely assess the postprandial mixed muscle fractional synthesis rate (FSR)—the definitive metric denoting the speed at which newly absorbed amino acids are structurally incorporated into skeletal myofibrillar proteins.
A single night of total sleep deprivation reduced the muscle protein fractional synthesis rate by a statistically significant 18 percent (decreasing from $0.072 \pm 0.015 \, \% \cdot h^{-1}$ under control conditions to $0.059 \pm 0.014 \, \% \cdot h^{-1}$ under sleep-deprived conditions; $P = 0.040$). This synthetic failure occurred simultaneously with severe shifts in the circulating endocrine environment: plasma cortisol (a potent catabolic glucocorticoid) increased by 21 percent ($P = 0.030$), while plasma testosterone (a primary anabolic androgen) plummeted by 24 percent ($P = 0.029$). Interestingly, circulating levels of insulin-like growth factor 1 (IGF-1) remained largely unchanged during this acute window.
| Muscle Metabolism Metric | Normal Sleep Control | Sleep Deprivation Intervention | Clinical Consequence |
| Fractional Synthesis Rate (FSR) | $0.072 \pm 0.015 \, \% \cdot h^{-1}$ | $0.059 \pm 0.014 \, \% \cdot h^{-1}$ | 18% reduction in the rate of generating new muscle proteins. |
| Plasma Cortisol | Baseline | +21% relative increase | Amplification of systemic stress and catabolic signaling. |
| Plasma Testosterone | Baseline | -24% relative decrease | Diminished androgenic support for tissue repair. |
| Proteolytic Gene Expression | Baseline | No significant acute change | Atrophy is driven primarily by synthetic failure, not immediate active degradation. |
Quantification of myofibrillar protein synthesis and hormonal shifts following acute sleep deprivation.
Perhaps the most illuminating finding from this transcriptomic profiling is that, while anabolic processes were severely blunted, the molecular markers of muscle protein degradation did not change acutely in response to the sleep deprivation. Specifically, muscle biopsies collected in the postprandial state revealed no alterations in the mRNA expression levels of the E3 ubiquitin ligases MuRF1 (muscle RING-finger protein-1) and MAFbx (muscle atrophy F-box, also known as atrogin-1 or FBOX-32), nor were there changes in the FOXO1 and FOXO3 transcription factors, or the microtubule-associated protein LC3. This suggests a highly nuanced pathophysiological sequence: sleep loss initiates clinical muscle atrophy not by immediately upregulating proteolytic destruction pathways (the ubiquitin-proteasome system), but by completely paralyzing the tissue’s ability to rebuild, repair, and remodel itself following daily mechanical and metabolic stress.
The Molecular Architecture of Synthetic Failure: The mTORC1-4EBP2 Axis
The exact molecular architecture governing this profound synthetic paralysis has been meticulously elucidated through in vivo protein translation assays, utilizing animal models to map the overlapping pathways between cognitive decline and muscular atrophy. Protein synthesis, whether in hippocampal neurons to facilitate the consolidation of long-term spatial memory or in skeletal muscle to facilitate myofibrillar hypertrophy, relies heavily on the insulin signaling pathway, including 5′ adenosine monophosphate-activated protein kinase (AMPK) and the mammalian target of rapamycin (mTOR).
mTOR is a serine/threonine kinase that forms a critical regulatory complex called mTOR complex 1 (mTORC1) when it associates with the regulatory-associated protein of mTOR (Raptor). mTORC1 acts as the primary cellular nutrient sensor; when energy is abundant and the cellular environment is permissive, mTORC1 stimulates downstream protein synthesis. It achieves this specifically by phosphorylating, and thereby inhibiting, the eukaryotic translation initiation factor 4E binding protein 2 (4EBP2).
Research indicates that sleep loss directly and aggressively attenuates the mTORC1-mediated phosphorylation of 4EBP2. When 4EBP2 remains unphosphorylated (due to the suppressed mTORC1 activity associated with sleep deprivation), it strongly binds to the eukaryotic initiation factor 4E (eIF4E). This binding physically prevents eIF4E from interacting with eukaryotic initiation factor 4G (eIF4G). The formation of the eIF4E-eIF4G complex is the absolute rate-limiting step for de novo protein production in the cell. By inhibiting this structural interaction, sleep deprivation effectively halts the assembly of the translation initiation machinery at the ribosomal level.
The causality of this specific molecular axis is so absolute that researchers found that artificially increasing the abundance of 4EBP2 in hippocampal excitatory neurons prior to sleep deprivation successfully increased the abundance of phosphorylated 4EBP2, fully restored the eIF4E-eIF4G interaction, and completely prevented the hippocampus-dependent memory deficits typically associated with sleep loss. Further studies subjecting subjects to intense treadmill exercise followed by 24 hours of REM sleep deprivation (REM-SD) confirmed that severe sleep restriction dramatically decreases the basal levels of mTOR expression in both the prefrontal cortex and the hippocampus, demonstrating that the translation-inhibiting effects of sleep loss are systemic and affect both cognitive and physical adaptation identically.
Mitigating Anabolic Resistance: High-Intensity Interval Exercise as a Countermeasure
While chronic sleep restriction (such as maintaining a schedule of only 4 hours in bed each night) chronically suppresses myofibrillar protein synthesis, therapeutic interventions can be strategically deployed to circumvent this suppression at the tissue level. An exhaustive parallel-group study was conducted to investigate the transcriptomic changes in human skeletal muscle following prolonged sleep restriction, and whether specific modalities of exercise could rescue the anabolic response.
Twenty-four healthy, young men were randomly allocated into three matched groups following two nights of controlled baseline sleep. The intervention lasted five nights. The first group, acting as the normal sleep (NS) control, was afforded 8 hours of time in bed (TIB) each night. The second group, the sleep restriction (SR) group, was limited to 4 hours TIB each night. The third group, the sleep restriction plus exercise (SR+EX) group, was also limited to 4 hours TIB each night, but importantly, they performed three sessions of High-Intensity Interval Exercise (HIIE) during the five-day period.
Total sleep time (TST) was significantly depressed in both restricted groups compared to the control (averaging 450 minutes for the NS group, 230 minutes for the SR group, and 234 minutes for the SR+EX group). Isotopic tracer analysis (deuterium oxide ingestion) revealed that the chronic sleep restriction group suffered a profound depression in their skeletal muscle fractional synthetic rate. The NS control group maintained a robust MyoPS rate of $1.53 \pm 0.09 \, \% \cdot day^{-1}$. The SR group saw this rate plummet to $1.24 \pm 0.21 \, \% \cdot day^{-1}$.
However, the SR+EX group, despite suffering from the identical severe sleep deprivation as the SR group, maintained an elevated MyoPS rate of $1.61 \pm 0.14 \, \% \cdot day^{-1}$, a level statistically indistinguishable from, and nominally higher than, the fully rested control group. This rescue effect suggests that the intense mechanical transduction and localized metabolic stress induced by high-intensity interval exercise can successfully bypass the systemic anabolic resistance caused by sleep loss. The intense muscular contractions likely upregulate mTOR pathway signaling independent of the systemic endocrine environment, providing a potent, non-pharmacological countermeasure to maintain myofibrillar remodeling and prevent the catastrophic degradation of skeletal muscle in chronically sleep-deprived populations.
Hedonic Drive, Neurobehavioral Alterations, and the Endocannabinoid System
The metabolic consequences of sleep restriction are drastically compounded by severe neurobehavioral alterations that heavily drive individuals toward caloric surpluses and exceptionally poor macronutrient selection. In free-living populations outside of highly controlled clinical trials, sleep deprivation shifts the locus of human feeding behavior away from homeostatic energy requirements (eating to sustain baseline metabolic function) and toward hedonic reward centers (eating for neurological pleasure), largely through the dysregulation of the endogenous cannabinoid system.
Endocannabinoids are naturally synthesized, endogenous lipid mediators that bind to the identical CB1 receptors targeted by the active psychoactive compounds in marijuana. Activation of these specific neural receptors heavily influences appetite regulation, mood stabilization, and reward-seeking behavior. The most abundant circulating endocannabinoid in the human bloodstream is 2-arachidonoylglycerol (2-AG).
Under normal, well-rested physiological conditions, circulating concentrations of 2-AG follow a robust and highly predictable circadian rhythm. Blood levels reach their nadir during the middle of the overnight fasting and sleep period, steadily climb throughout the morning, and typically peak in the early afternoon (around 12:30 p.m.) before gradually declining throughout the late afternoon and evening. However, when sleep is restricted, the entire daily rhythm of 2-AG is severely amplified and distorted. Sleep-restricted individuals exhibit delayed, vastly extended, and significantly higher maximum 2-AG values that remain elevated well into the evening—the exact time of day when mindless snacking is most strongly linked to pathological weight gain.
This massive amplification of the endocannabinoid system produces a distinct and overwhelming behavioral phenotype. Concomitant with the afternoon and evening elevations of 2-AG, sleep-restricted individuals report dramatic increases in subjective hunger, cravings, and a specific, intense desire to eat highly palatable, reward-dense foods (specifically snacks high in refined carbohydrates and fats, such as cookies, candy, and chips).
In clinical observations, sleep-deprived participants were unable to cognitively inhibit the intake of these hyperpalatable snacks, despite the fact that they had consumed a massive, nutritionally complete meal that supplied 90 percent of their total daily caloric needs just two hours prior. The biological urge superseded cognitive restraint. When granted ad libitum access to snacks, these individuals consumed nearly twice as much dietary fat compared to when they had obtained a full eight hours of restorative sleep.
This phenomenon is severely exacerbated by simultaneous disruptions in traditional, homeostatic satiety hormones. Short-term sleep restriction (such as four hours in bed for two consecutive nights) results in a 24 percent increase in circulating ghrelin (the stomach-derived hormone that stimulates hunger and reduces energy expenditure) and an 18 percent decrease in leptin (the adipocyte-derived hormone that signals long-term satiety to the hypothalamus). The convergence of abnormally low leptin, pathologically high ghrelin, and a massively overactive endocannabinoid system creates an overwhelming neurobiological drive to overeat. The elevated 2-AG levels increase the “hedonic aspect of food intake,” effectively magnifying the neurological pleasure, satisfaction, and dopamine release gained from consuming hypercaloric foods. Consequently, the probability of a human subject adhering to a strict caloric deficit while chronically sleep-deprived approaches zero, fundamentally undermining dietary interventions for weight management from a psychological and behavioral standpoint.
The Endocrine Milieu: Testosterone, Cortisol, and Growth Hormone Dynamics
The systemic disruption of substrate utilization, cellular signaling, and neurobehavioral restraint is ultimately orchestrated by the severe derangement of the body’s primary hormonal axes. Sleep is the foundational regulatory period for the secretion and modulation of testosterone, cortisol, and growth hormone (GH), and the precise temporal timing of their release is as critical to human physiology as the absolute volume secreted.
The balance between anabolic hormones (which build tissue) and catabolic hormones (which degrade tissue)—specifically the ratio of testosterone to cortisol—is a paramount determinant of tissue adaptation, immunological function, and athletic performance. Cortisol, the primary glucocorticoid produced by the adrenal glands, plays a central role in the physiological and behavioral response to stress, managing glucose regulation and immune suppression. Typically, cortisol follows a strict diurnal pattern: it peaks in the early morning hours to facilitate wakefulness and gradually declines to its lowest levels at night to allow for cellular repair.
However, recurrent short sleep duration, as observed in epidemiological cohorts of thousands of adults, is strongly associated with higher late-afternoon and evening salivary cortisol concentrations. This altered temporal presentation is metabolically devastating; sustained high levels of evening cortisol induce profound insulin resistance the following morning, perpetuating the exact adipocyte dysfunction discussed previously, and promoting the central accumulation of visceral belly fat.
Conversely, testosterone, a steroidal androgen associated with muscular hypertrophy, central nervous system recovery, and red blood cell production, is deeply reliant on sleep for its production and pulsatile release. Clinical studies utilizing frequent, intensive blood sampling methodologies demonstrate that sleep restriction unequivocally decreases systemic 24-hour testosterone concentrations. Sleep restriction not only lowers morning testosterone baselines by up to 24 percent but also severely disrupts the pulsatile secretion frequency, particularly in older populations. The reduction in luteinizing hormone (LH) pulse mass during the morning hours directly limits the gonadal synthesis of testosterone, establishing a systemic, albeit temporary, hypogonadal state that limits muscular hypertrophy, reduces peak power output, slows reaction times, and delays neurological recovery from intense physical exertion.
| Endocrine Marker | Normal Sleep Function | Pathological Consequence of Sleep Restriction | Direct Metabolic Implication |
| Testosterone | Pulsatile release governed by overnight LH spikes; primary driver of MyoPS. | Up to 24% acute reduction; severely disrupted pulse frequency and mass. | Anabolic resistance; failure to recover from mechanical muscular stress; reduced power output. |
| Cortisol | Morning peak for wakefulness; evening nadir for cellular repair and insulin sensitivity. | Elevated afternoon/evening concentrations; 21% acute overall daily increase. | Promotion of systemic insulin resistance; active breakdown of fat-free mass; visceral fat storage. |
| Growth Hormone | Massive surge during the earliest phases of slow-wave (deep) sleep; highly lipolytic. | Severely blunted nocturnal surge due to truncated deep sleep architecture. | Diminished lipid oxidation; impaired skeletal tissue regeneration; impaired injury recovery. |
Summary of primary endocrine shifts corresponding to sleep deprivation and their metabolic consequences.
Furthermore, Growth Hormone (GH) is uniquely and inextricably tied to the sleep cycle. The vast majority of the body’s daily GH secretion occurs in massive, concentrated pulses during the earliest phases of slow-wave (deep) sleep. GH is highly lipolytic and strongly muscle-sparing; an extended exposure of peripheral tissues to sufficient GH levels dictates structural recovery, tissue repair, and the mobilization of stored fats.
When sleep duration is curtailed, the duration of deep sleep is disproportionately truncated, directly blunting the nocturnal surge of GH. This effectively shortchanges the human body of its most potent endogenous tissue-repair mechanism. Furthermore, disrupted GH production leads to accelerated fat accumulation and severely decreased energy levels during waking hours. Without the powerful lipolytic action of GH, the body loses a critical mechanism for breaking down triglycerides, further exacerbating the pathological fat retention observed in the hypocaloric sleep-deprivation trials.
Objective Quantification in Sports Science: Wearable Biometrics and Autonomic Proxies
Given the severe, systemic metabolic and endocrine consequences of sleep restriction, the objective quantification of sleep architecture and physiological recovery has transitioned from a clinical curiosity to a mandatory performance metric in elite sports science, bodybuilding, and human performance optimization. The integration of continuous physiological monitoring allows for the empirical management of an athlete’s recovery status, substituting subjective guesswork with hard, actionable data.
The undisputed gold standard for assessing non-invasive systemic recovery is Heart Rate Variability (HRV), a biomarker that reflects the dynamic interplay and balance of the autonomic nervous system. Specifically, the root mean square of successive differences (RMSSD) is utilized to track parasympathetic (rest and digest) activity. Sleep deprivation, by elevating cortisol and shifting the body into a state of perceived crisis, drastically reduces parasympathetic tone while upregulating sympathetic (fight or flight) activity, manifesting as a severely suppressed RMSSD. When an athlete experiences incomplete sleep, the sympathetic dominance suppresses the heart’s natural beat-to-beat variability. Elite athletes displaying a downward 7-day rolling average in their HRV metric are functionally exhibiting central nervous system fatigue, heavily indicating that the endocrine environment is skewed toward catabolism and that intense training will likely yield injury rather than adaptation.
Modern clinical applications and elite sports science heavily rely on sophisticated wearable biometric devices (such as Oura, Whoop, Garmin, and Ultrahuman rings) to track these vital parameters. While absolute sleep staging accuracy (the ability of a wrist or finger wearable to perfectly delineate light, deep, and REM sleep based on movement and heart rate) ranges from 51 percent to 65 percent across devices, the long-term trend analysis is exceptionally robust and highly actionable. A systematic review of clinical sleep monitoring notes that wearables are invaluable for tracking these physiological patterns over time.
A critical finding from massive longitudinal data sets—including a 2025 study analyzing nearly a million days of user data from 11,914 Whoop users—reveals that sleep consistency (the strict regularity of bedtimes and wake times) is as important, if not more so, than total sleep duration. High sleep consistency correlates with significantly lower resting heart rates, higher baseline HRV, and a highly stabilized endocrine response. For athletes, tracking deviations in metrics such as the overnight resting heart rate (RHR) or respiratory rate provides immediate feedback. An elevated overnight RHR relative to a user’s baseline, even if the athlete perceives themselves as subjectively rested, is a primary indicator of incomplete recovery and lingering sympathetic arousal. To optimize recovery, sports scientists dictate that total sleep duration should fall between 7.5 and 9 hours, with deep sleep comprising 15 to 20 percent of the total, REM sleep comprising 20 to 25 percent, and overall sleep efficiency remaining above 80 percent.
Clinical and Athletic Implications: Periodization of Nutrition and Training
The cascade of physiological failures initiated by sleep restriction—ranging from profound adipocyte insulin resistance and suppressed mTORC1 signaling to the hyperactivation of the endocannabinoid system and the suppression of the testosterone-to-cortisol ratio—demands highly structured corrective interventions for anyone attempting to alter their body composition.
The most vital clinical realization is that nutritional interventions alone cannot rescue the metabolic damage inflicted by sleep deprivation. Because the loss of sleep inherently biases the body’s nutrient partitioning away from lipid oxidation and toward the rapid retention of fat and the oxidation of structural amino acids, administering a hypocaloric diet during periods of sleep restriction will selectively destroy skeletal muscle. One simply cannot out-diet the endocrine consequences of sleep deprivation.
Therefore, dietary and training periodization must strictly account for sleep load. During phases of unavoidable sleep restriction (e.g., due to newborn care, occupational shifts, or high academic/corporate stress), caloric deficits should be entirely abandoned in favor of maintenance calories. Within this maintenance phase, macronutrient distribution must heavily favor high protein intakes to leverage the thermic effect of food and provide an abundance of circulating exogenous amino acids to combat the elevated rates of muscle protein breakdown. Furthermore, implementing high-intensity interval training, rather than long-duration steady-state cardio which may further elevate cortisol, acts as a physiological wedge, bypassing the sleep-induced blockades on myofibrillar protein synthesis and mitigating the loss of fat-free mass.
Conclusion
The characterization of sleep as a passive biological necessity severely underestimates its role as the absolute primary governor of human metabolism. Exhaustive clinical data unequivocally demonstrates that sleep architecture dictates the systemic hormonal environment, which in turn commands cellular nutrient partitioning at the molecular level. When sleep is restricted, the human body interprets the extended wakefulness and accompanying cortisol elevations as a severe, existential survival threat. It responds by inducing profound insulin resistance at the level of the adipocyte, effectively locking lipid stores and driving compensatory hyperinsulinemia. Simultaneously, the suppression of the mTORC1-4EBP2 axis paralyzes the synthesis of new muscle proteins, forcing the body to aggressively catabolize its own structural tissues to meet circulating energy demands.
Furthermore, the pathological amplification of the endocannabinoid system, specifically the dramatic elevations of 2-arachidonoylglycerol, coupled with derangements in leptin, ghrelin, cortisol, testosterone, and growth hormone, creates a neuroendocrine environment that aggressively drives the overconsumption of hyperpalatable foods while completely sabotaging tissue recovery. For individuals engaged in weight loss, physique recomposition, or elite athletic pursuits, sleep cannot be viewed as a secondary or auxiliary lifestyle factor. It is the fundamental physiological prerequisite that allows the body to safely oxidize fat, synthesize functional muscle tissue, and adapt to physical stress. Failing to quantify, protect, and optimize sleep as a core performance metric guarantees suboptimal physiological outcomes, rendering even the most meticulously designed nutritional and resistance training interventions biologically ineffective.